Preprocessing Tutorial
This tutorial is designed to show you how Dynamo preprocess the regular
scRNA-seq or time-resolved metabolic labeling enabled scRNA-seq data. In
this new release of dynamo 1.3.0, we introduce the Preprocessor
class (many parts of it has been implemented previously and but
extensively updated and refined in this release), which allows you to
convenietntly explore different preprocessing recipes with configurable
parameters inside the class. The Preprocessor currently includes
several existing recipes such as monocle, pearson residual,
seurat, and sctransform. Furthermore, you can easily replace
each preprocessing step with your own method of preprocessing. For
example, the Preprocessor’s monocle pipeline contains steps such as
filter_cells_by_outliers, filter_genes_by_outliers,
normalize_by_cells, and select_genes. You can replace the
default preprocessing steps with your own method and modify default
monocle parameters passed into these functions by reconfiguring the
attributes of the Preprocessor class.
Here is a flowchart depicting the structure of the preprocessing module in Dynamo 1.3. It lists all python files and main functions implemented in the preprocessing module. By clicking on a block (e.g. normalization), you will be directed to the corresponding file, and clicking on a specific API (e.g. calc_sz_factor) will take you to the associated functions.
The recipes in dynamo
To make your life easy, currently dynamo supports 4 major recipe for preprocessing. Please find at the end of this tutorial how you can customize your preprocessing method.
Monocle: Monocle recipe uses similar but generalized strategy from Monocle 3 to normalize all datasets in different layers (the spliced and unspliced or new, i.e. metabolic labelled, and total mRNAs or others), followed by feature selection, log1p transformation of the data and principal component analysis (PCA) dimension reduction. For more details, please refer to the paper and code.Pearson Residuals: Pearson Residuals recipe implements a preprocessing pipeline proposed in the study by Lause, Berens, and Kobak (2021) that uses Pearson Residuals for the normalization of single-cell RNA-seq UMI data. This method performs several steps including standardization, filtering of cells and genes by outliers, select_genes_by_pearson_residuals, appending or excluding gene lists, normalize_layers_pearson_residuals, regression, and PCA. This pipeline aims to preprocess the data using the Pearson residuals method to prepare it for downstream analysis. For more details, please refer to the paper and code.Seurat: Seurat recipe implements a preprocessing pipeline based on the Seurat package’s approach to single-cell data integration and analysis. This recipe performs various steps including standardization, filtering of cells and genes by outliers, calculation of size factors, normalization by cells, gene selection, appending or excluding gene lists, normalization method selection, regression, and PCA. This pipeline follows the Seurat methodology for preprocessing single-cell data, as described in the publications by Stuart and Butler et al. (Cell, 2019) and Butler et al. (Nat Biotechnol). The goal is to prepare the data for downstream analysis and integration using Seurat’s methods. For more details, please refer to the code and documentation.Sctransform: Sctransform recipe implements a preprocessing pipeline based on the sctransform method developed by Hao and Hao et al. This performs several steps including standardization, filtering of cells and genes by outliers, gene selection, appending or excluding gene lists, performing the sctransform method, regression, and PCA. The sctransform method is a model of single cell UMI expression data using regularized negative binomial regression and an integrated analysis approach for multimodal single-cell data. This pipeline aims to preprocess the data using the sctransform method to account for downstream analysis. For more details, please refer to the paper, code, and R code.
Note: In older versions, dynamo offered several recipes, among which
recipe_monocle is a basic function as a building block of other
recipes. You can still use these functions to preprocess data.
Preprocessor provides users with config_monocle_recipe and other
config_*_recipes methods to help you reproduce different
preprocessor results and integrate with your newly developed
preprocessing algorithms.
Without further ado, let’s begin by importing packages.
import warnings
warnings.filterwarnings('ignore')
warnings.filterwarnings("ignore", message="numpy.dtype size changed")
import dynamo as dyn
import seaborn as sns
import matplotlib.pyplot as plt
import matplotlib
import numpy as np
If you would like to enable debug log messages to check the preprocessing procedure, simply uncomment the code snippet below
# dyn.dynamo_logger.main_set_level("DEBUG")
Glossary of keys generated during preprocessing
you do need to update all list to series…
adata.obs.pass_basic_filter: a series of boolean variables indicating whether cells pass certain basic filters. In monocle recipe, the basic filtering is based on thresholding of expression values.adata.var.pass_basic_filter: a series of boolean variables indicating whether genes pass certain basic filters. In monocle recipe, the basic filtering is based on thresholding of expression values.adata.var.use_for_pca: a series of boolean variables points to feature genes selected for PCA dimension reduction and following downstream RNA velocity and vector field analysis. In many recipes, this key is equivalent to highly variable genes.adata.var.highly_variable_scores: a series of float number scores indicating how variable each gene is, typically generated during gene feature selection (preprocessor.select_genes). Note only part of recipes do not have this highly variable scores. E.g.seuratV3recipe implemented in dynamo does not have highly variable scores due to its thresholding nature.adata.layers['X_spliced']: spliced expression matrix after normalization used in downstream computation.data.layers['X_unspliced']: unspliced expression matrix after normalization used in downstream computation.adata.obsm['X_pca']: normalized X after PCA transformation.adata.X: normalized X (e.g. size factor normalized and log1p transformed)
Using predefined (default) recipe configurations in preprocessor
Firstly, you can just start with the inclusion of a preprocessor, so that your life can become much easier. With just three lines of code, the preprocessor can handle the entire process of data filtering, manipulation, calculation, and conversion. You no longer have to worry about the headaches associated with these tasks.
from dynamo.preprocessing import Preprocessor
# download the data
adata = dyn.sample_data.zebrafish()
celltype_key = "Cell_type"
|-----> Downloading data to ./data/zebrafish.h5ad
adata
AnnData object with n_obs × n_vars = 4181 × 16940
obs: 'split_id', 'sample', 'Size_Factor', 'condition', 'Cluster', 'Cell_type', 'umap_1', 'umap_2', 'batch'
layers: 'spliced', 'unspliced'
preprocessor = Preprocessor()
preprocessor.preprocess_adata(adata, recipe="monocle")
# Alternative
# preprocessor.config_monocle_recipe(adata)
# preprocessor.preprocess_adata_monocle(adata)
|-----> Running monocle preprocessing pipeline...
|-----------> filtered out 14 outlier cells
|-----------> filtered out 12746 outlier genes
|-----> PCA dimension reduction
|-----> <insert> X_pca to obsm in AnnData Object.
|-----> [Preprocessor-monocle] completed [3.5468s]
adata
AnnData object with n_obs × n_vars = 4167 × 16940
obs: 'split_id', 'sample', 'Size_Factor', 'condition', 'Cluster', 'Cell_type', 'umap_1', 'umap_2', 'batch', 'nGenes', 'nCounts', 'pMito', 'pass_basic_filter', 'initial_cell_size', 'unspliced_Size_Factor', 'initial_unspliced_cell_size', 'spliced_Size_Factor', 'initial_spliced_cell_size', 'ntr'
var: 'nCells', 'nCounts', 'pass_basic_filter', 'log_cv', 'score', 'log_m', 'frac', 'use_for_pca', 'ntr'
uns: 'pp', 'velocyto_SVR', 'feature_selection', 'PCs', 'explained_variance_ratio_', 'pca_mean'
obsm: 'X_pca'
layers: 'spliced', 'unspliced', 'X_spliced', 'X_unspliced'
dyn.tl.reduceDimension(adata, basis="pca")
# pointsize can be used to set the size of data points (cells) while alpha set the transparency value of the data points
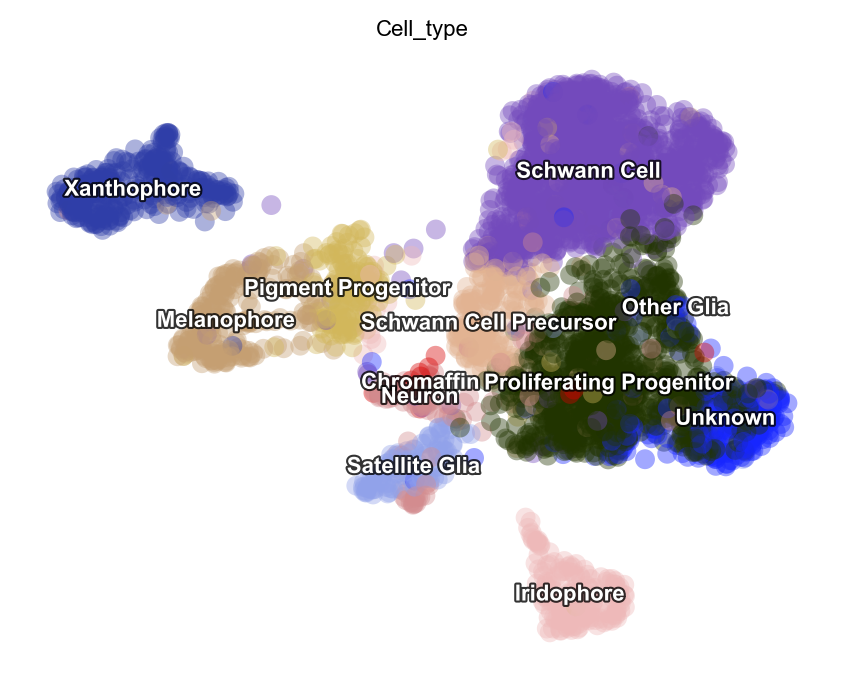
dyn.pl.umap(adata, color=celltype_key, pointsize=0.2, alpha=0.4)
|-----> retrieve data for non-linear dimension reduction...
|-----> [UMAP] using X_pca with n_pca_components = 30
|-----> <insert> X_umap to obsm in AnnData Object.
|-----> [UMAP] completed [27.4297s]
|-----------> plotting with basis key=X_umap
|-----------> skip filtering Cell_type by stack threshold when stacking color because it is not a numeric type
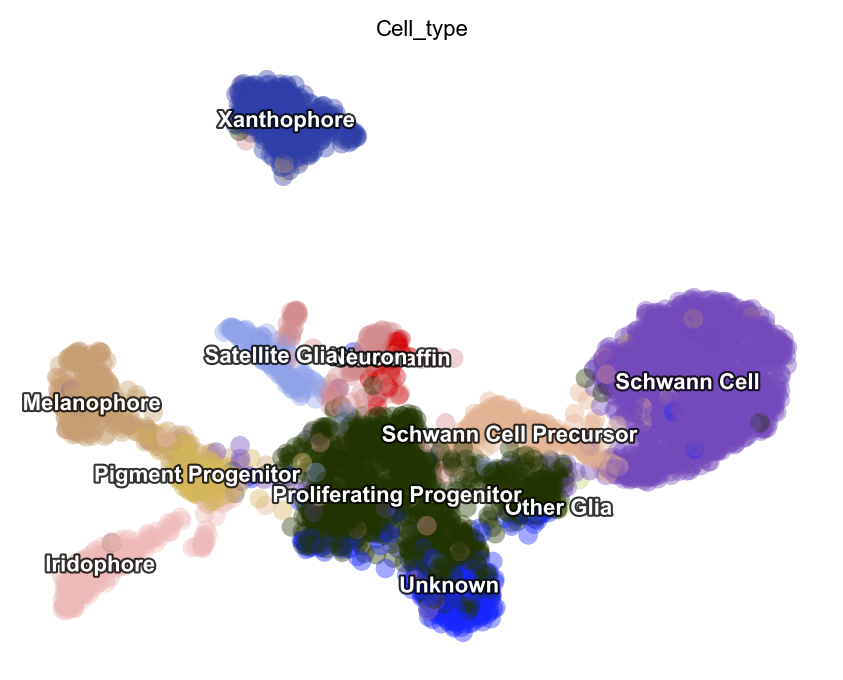
Please note that pearson residual or sctransform transformation should only be performed for adata.X and not applied to different layers. This is because RNA velocity do have physical meanings, and otherwise spliced/unspliced data will reseult in negative values after transformation.
adata = dyn.sample_data.zebrafish()
preprocessor = Preprocessor()
preprocessor.preprocess_adata(adata, recipe="pearson_residuals")
# Alternative
# preprocessor.config_pearson_residuals_recipe(adata)
# preprocessor.preprocess_adata_pearson_residuals(adata)
|-----> Downloading data to ./data/zebrafish.h5ad
|-----> gene selection on layer: X
|-----> extracting highly variable genes
|-----------> filtered out 350 outlier genes
|-----> applying Pearson residuals to layer <X>
|-----> replacing layer <X> with pearson residual normalized data.
|-----> [pearson residual normalization for X] completed [1.1042s]
|-----> PCA dimension reduction
|-----> <insert> X_pca to obsm in AnnData Object.
|-----> [Preprocessor-pearson residual] completed [4.8708s]
dyn.tl.reduceDimension(adata)
dyn.pl.umap(adata, color=celltype_key, pointsize=0.2, alpha=0.4)
|-----> retrieve data for non-linear dimension reduction...
|-----> [UMAP] using X_pca with n_pca_components = 30
|-----> <insert> X_umap to obsm in AnnData Object.
|-----> [UMAP] completed [16.7289s]
|-----------> plotting with basis key=X_umap
|-----------> skip filtering Cell_type by stack threshold when stacking color because it is not a numeric type
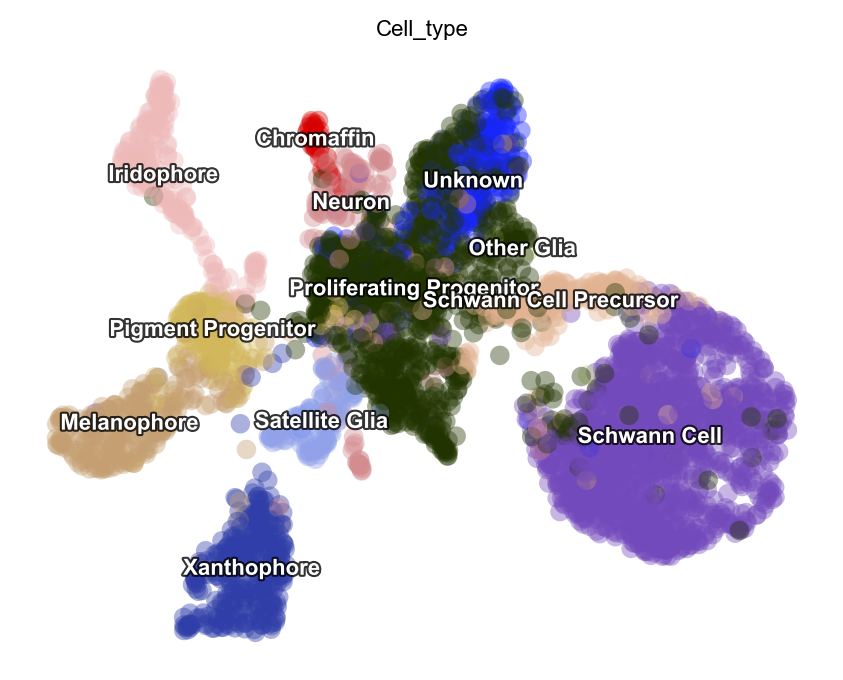
Sctransform transformation is only applied to adata.X and not applied to different layers, as stated above.
import warnings
warnings.filterwarnings('ignore', category=UserWarning, message='Сould not load pytorch')
warnings.filterwarnings("ignore", category=DeprecationWarning)
adata = dyn.sample_data.zebrafish()
preprocessor = Preprocessor()
preprocessor.preprocess_adata(adata, recipe="sctransform")
# Alternative
# preprocessor.config_sctransform_recipe(adata)
# preprocessor.preprocess_adata_sctransform(adata)
|-----> Downloading data to ./data/zebrafish.h5ad
|-----> Running Sctransform recipe preprocessing...
|-----------> filtered out 14 outlier cells
|-----------> filtered out 12410 outlier genes
|-----? Sctransform recipe will subset the data first with default gene selection function for efficiency. If you want to disable this, please perform sctransform without recipe.
|-----> sctransform adata on layer: X
|-----------> set sctransform results to adata.X
|-----> PCA dimension reduction
|-----> <insert> X_pca to obsm in AnnData Object.
|-----> [Preprocessor-sctransform] completed [16.6057s]
dyn.tl.reduceDimension(adata)
dyn.pl.umap(adata, color=celltype_key, pointsize=0.2, alpha=0.4)
|-----> retrieve data for non-linear dimension reduction...
|-----> [UMAP] using X_pca with n_pca_components = 30
|-----> <insert> X_umap to obsm in AnnData Object.
|-----> [UMAP] completed [16.2677s]
|-----------> plotting with basis key=X_umap
|-----------> skip filtering Cell_type by stack threshold when stacking color because it is not a numeric type
adata = dyn.sample_data.zebrafish()
preprocessor = Preprocessor()
# Alternative
# preprocessor.config_seurat_recipe(adata)
# preprocessor.preprocess_adata_seurat(adata)
preprocessor.preprocess_adata(adata, recipe="seurat")
|-----> Downloading data to ./data/zebrafish.h5ad
|-----> Running Seurat recipe preprocessing...
|-----------> filtered out 14 outlier cells
|-----------> filtered out 11388 outlier genes
|-----> select genes on var key: pass_basic_filter
|-----------> choose 2000 top genes
|-----> number of selected highly variable genes: 2000
|-----> PCA dimension reduction
|-----> <insert> X_pca to obsm in AnnData Object.
|-----> [Preprocessor-seurat] completed [1.4041s]
dyn.tl.reduceDimension(adata)
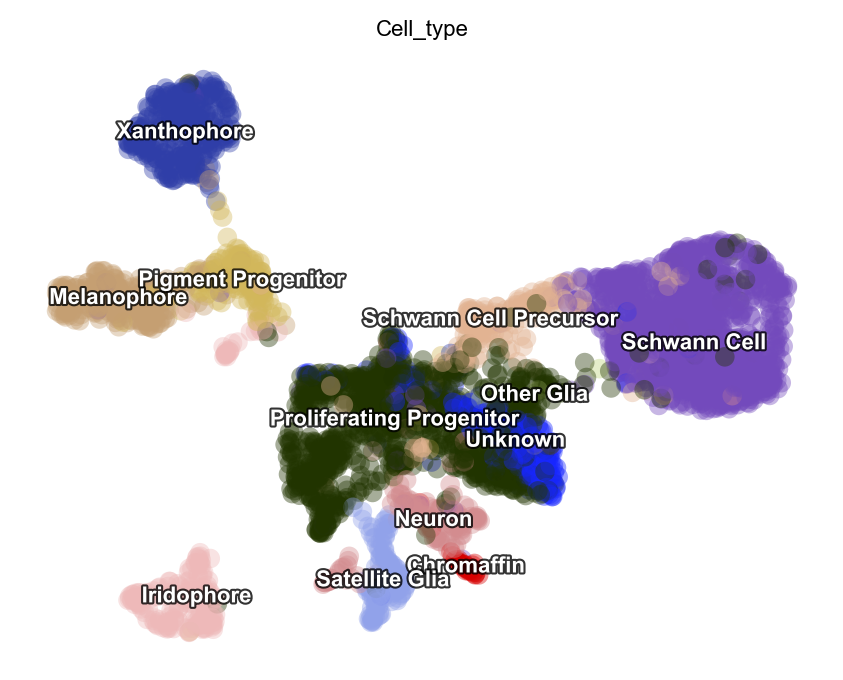
dyn.pl.umap(adata, color=celltype_key, pointsize=0.2, alpha=0.4)
|-----> retrieve data for non-linear dimension reduction...
|-----> [UMAP] using X_pca with n_pca_components = 30
|-----> <insert> X_umap to obsm in AnnData Object.
|-----> [UMAP] completed [16.0549s]
|-----------> plotting with basis key=X_umap
|-----------> skip filtering Cell_type by stack threshold when stacking color because it is not a numeric type
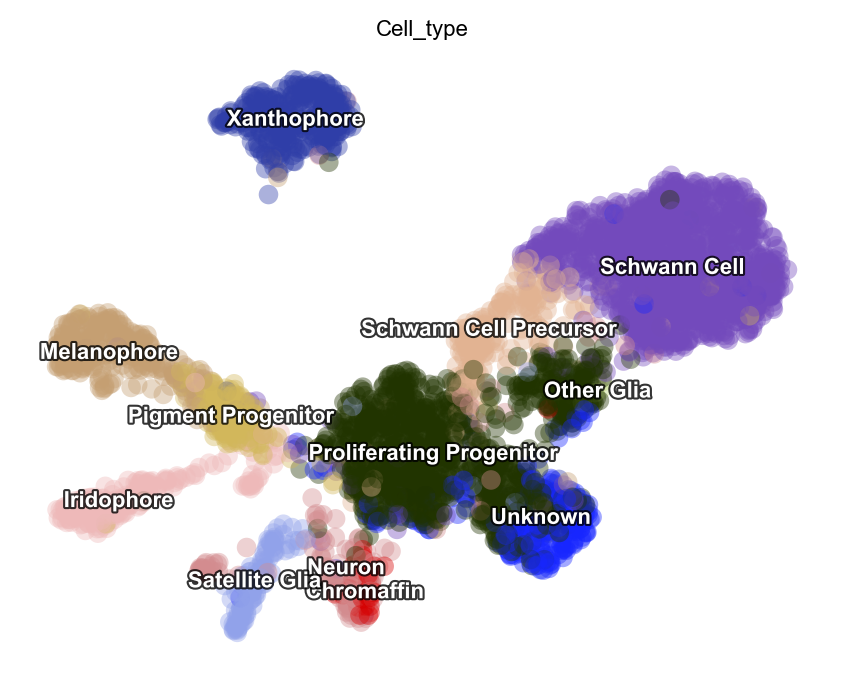
Customize function parameters configured in Preprocessor
In this example, we will use the monocle recipe to demonstrate how we
can configure Preprocessor cluster to select genes based on your needs.
Note that we can set the recipe to be dynamo_monocle, seurat, or
others to apply different criteria for selecting genes. We can also set
the select_genes_kwargs parameter in the preprocessor to pass
additional desired parameters. By default, the recipe is set to
dynamo_monocle. We can change it to seurat and add other
constraint parameters if needed.
adata = dyn.sample_data.zebrafish()
preprocessor = Preprocessor()
preprocessor.config_monocle_recipe(adata)
|-----> Downloading data to ./data/zebrafish.h5ad
preprocessor.select_genes_kwargs contains arguments that will be
passed to select_genes step.
preprocessor.select_genes_kwargs
{'n_top_genes': 2000, 'SVRs_kwargs': {'relative_expr': False}}
To set the preprocessing steps and their corresponding function
parameters for the monocle recipe, we can call
preprocessor.config_monocle_recipe(). By default, the constructor
parameters of the Preprocessor for preprocessing are set to the monocle
recipe used in report the Dynamo cell paper
dynamo.
If you would like to customize the dataset to better fit your preferences, you can adjust the parameters before running the recipe. Here is an example.
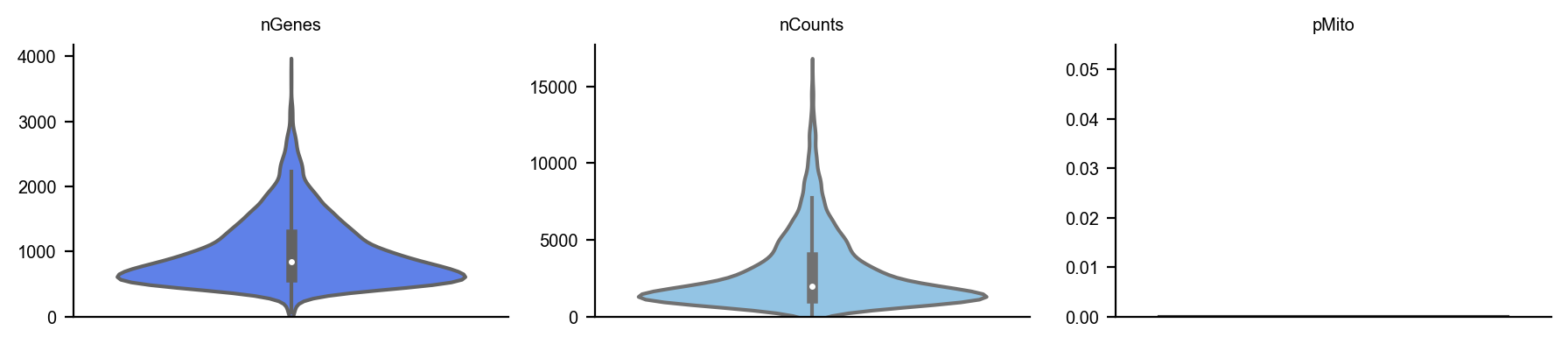
To begin, you can create a plot of the basic statistics (nGenes, nCounts, and pMito) for each category of adata. - nGenes: the number of genes - nCounts: the number of cells - pMito: the percentage of mitochondria genes.
dyn.pl.basic_stats(adata, figsize=(3, 2))
You can visualize the rank of the fraction of UMI to the total UMI per cell for the top 20 genes
dyn.pl.highest_frac_genes(adata, figsize=(6, 2))
|-----------? use_for_pca not in adata.var, ignoring the gene annotation key when plotting
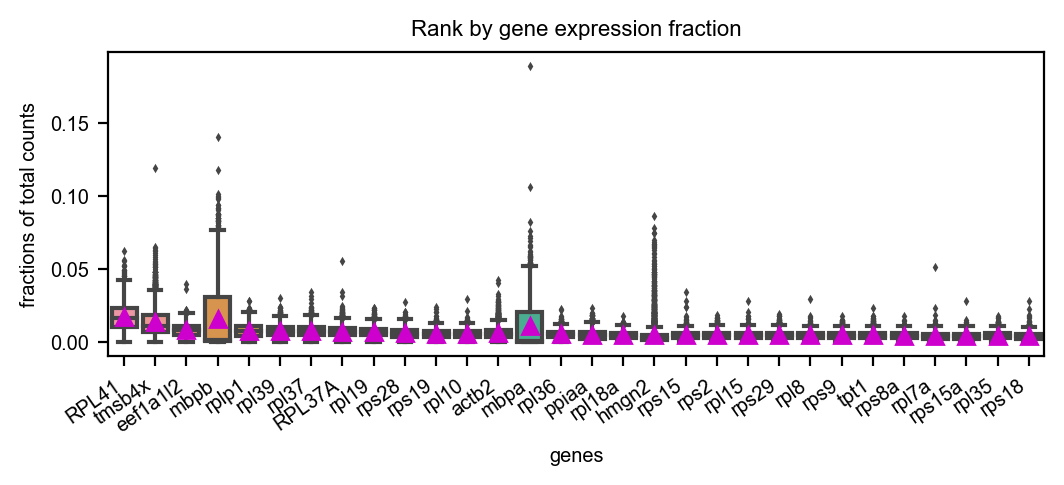
<AxesSubplot:title={'center':'Rank by gene expression fraction'}, xlabel='genes', ylabel='fractions of total counts'>
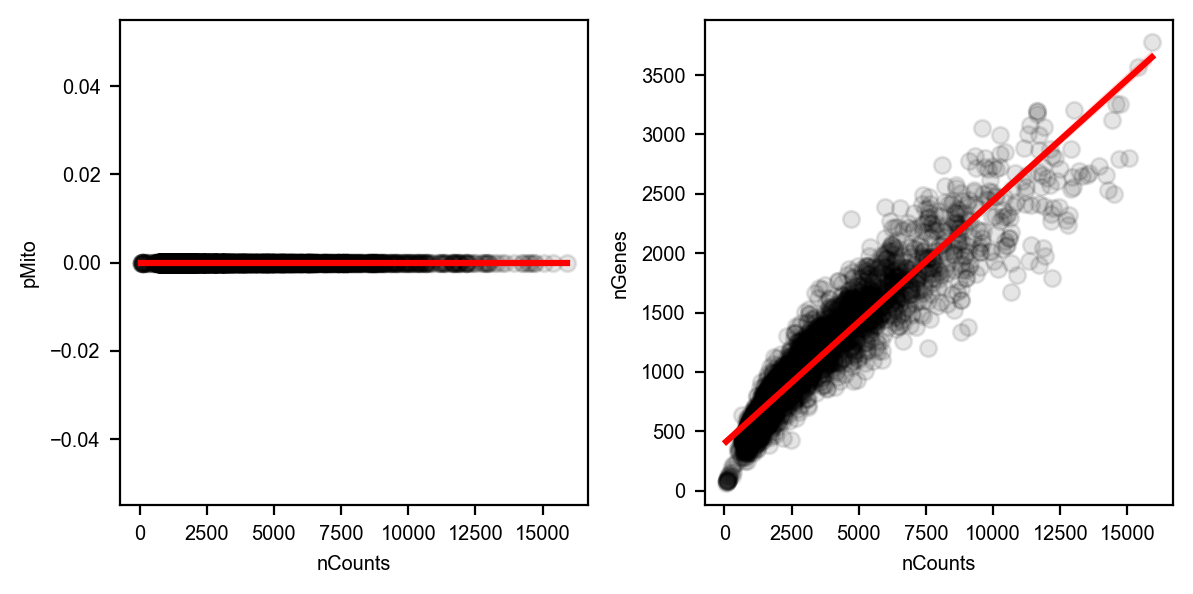
You can get rid of any cells that have mitochondrial gene expression percentage greater than pMito or total counts greater than nCounts. You can adjust the threshold values as per your requirements.
Note that in our particular case, our data doesn’t have mitochondria counts.
import seaborn as sns
import matplotlib.pyplot as plt
# Create the first figure
df = adata.obs.loc[:, ["nCounts", "pMito", "nGenes"]]
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(6, 3)) # Adjust the figsize as desired
sns.regplot(data=df,
x="nCounts",
y="pMito",
ax=ax1,
scatter_kws={"color": "black", "alpha": 0.1},
line_kws={"color": "red"})
ax1.set_xlabel("nCounts")
ax1.set_ylabel("pMito")
# Create the second figure
sns.regplot(data=df,
x="nCounts",
y="nGenes",
ax=ax2,
scatter_kws={"color": "black", "alpha": 0.1},
line_kws={"color": "red"})
ax2.set_xlabel("nCounts")
ax2.set_ylabel("nGenes")
# Display the figures side by side
plt.tight_layout() # Optional: Adjusts spacing between subplots
plt.show()
And modify some values of parameters based on the information above.
preprocessor.filter_cells_by_outliers_kwargs = {
"filter_bool": None,
"layer": "all",
"min_expr_genes_s": 300,
"min_expr_genes_u": 100,
"min_expr_genes_p": 50,
"max_expr_genes_s": np.inf,
"max_expr_genes_u": np.inf,
"max_expr_genes_p": np.inf,
"shared_count": None,
}
preprocessor.filter_genes_by_outliers_kwargs = {
"filter_bool": None,
"layer": "all",
"min_cell_s": 3,
"min_cell_u": 2,
"min_cell_p": 1,
"min_avg_exp_s": 0,
"min_avg_exp_u": 0,
"min_avg_exp_p": 0,
"max_avg_exp": np.inf,
"min_count_s": 5,
"min_count_u": 0,
"min_count_p": 0,
"shared_count": 40,
}
preprocessor.select_genes_kwargs = {
"n_top_genes": 2500,
"sort_by": "cv_dispersion",
"keep_filtered": True,
"SVRs_kwargs": {
"relative_expr": True,
"total_szfactor": "total_Size_Factor",
"min_expr_cells": 0,
"min_expr_avg": 0,
"max_expr_avg": np.inf,
"winsorize": False,
"winsor_perc": (1, 99.5),
"sort_inverse": False,
"svr_gamma": None,
},
}
Let`s run the monocle recipe again.
preprocessor.preprocess_adata_monocle(adata)
|-----> Running monocle preprocessing pipeline...
|-----------> filtered out 125 outlier cells
|-----------> filtered out 13035 outlier genes
|-----> PCA dimension reduction
|-----> <insert> X_pca to obsm in AnnData Object.
|-----> [Preprocessor-monocle] completed [2.9955s]
dyn.tl.reduceDimension(adata, basis="pca")
dyn.pl.umap(adata, color=celltype_key, pointsize=0.2, alpha=0.4)
|-----> retrieve data for non-linear dimension reduction...
|-----> [UMAP] using X_pca with n_pca_components = 30
|-----> <insert> X_umap to obsm in AnnData Object.
|-----> [UMAP] completed [25.0124s]
|-----------> plotting with basis key=X_umap
|-----------> skip filtering Cell_type by stack threshold when stacking color because it is not a numeric type
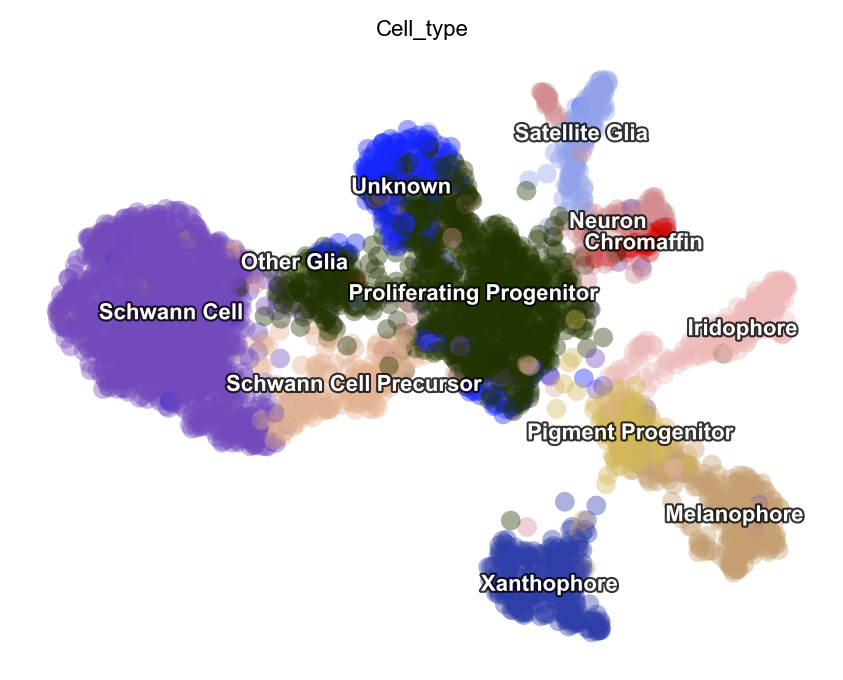
Let`s run the seurat recipe in this time.
adata = dyn.sample_data.zebrafish()
preprocessor = Preprocessor()
preprocessor.config_seurat_recipe(adata)
|-----> Downloading data to ./data/zebrafish.h5ad
preprocessor.select_genes_kwargs
{'algorithm': 'seurat_dispersion', 'n_top_genes': 2000}
preprocessor.select_genes_kwargs = dict(
n_top_genes=2500,
algorithm="seurat_dispersion", # or "fano_dispersion"
seurat_min_disp=None,
seurat_max_disp=None,
seurat_min_mean=0.4,
seurat_max_mean=0.6,
)
preprocessor.select_genes_kwargs
{'n_top_genes': 2500,
'algorithm': 'seurat_dispersion',
'seurat_min_disp': None,
'seurat_max_disp': None,
'seurat_min_mean': 0.4,
'seurat_max_mean': 0.6}
preprocessor.preprocess_adata_seurat(adata)
dyn.tl.reduceDimension(adata, basis="pca")
dyn.pl.umap(adata, color=celltype_key, pointsize=0.2, alpha=0.4)
|-----> Running Seurat recipe preprocessing...
|-----------> filtered out 14 outlier cells
|-----------> filtered out 11388 outlier genes
|-----> select genes on var key: pass_basic_filter
|-----------> choose 2500 top genes
|-----> number of selected highly variable genes: 2500
|-----> PCA dimension reduction
|-----> <insert> X_pca to obsm in AnnData Object.
|-----> [Preprocessor-seurat] completed [1.4892s]
|-----> retrieve data for non-linear dimension reduction...
|-----> [UMAP] using X_pca with n_pca_components = 30
|-----> <insert> X_umap to obsm in AnnData Object.
|-----> [UMAP] completed [15.9357s]
|-----------> plotting with basis key=X_umap
|-----------> skip filtering Cell_type by stack threshold when stacking color because it is not a numeric type
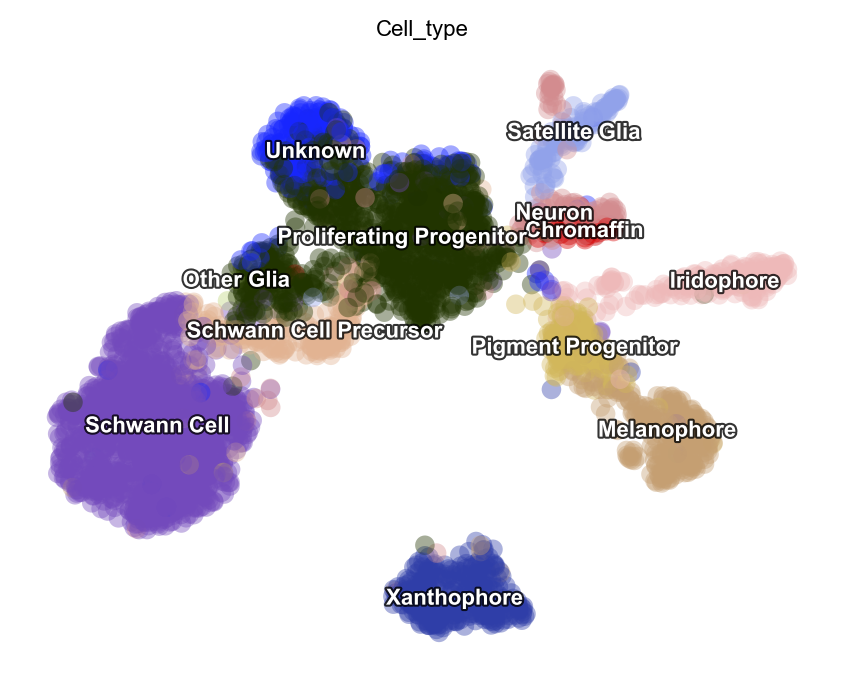
Customize and run each functions directly.
We understand that some of you may prefer to use the each function by calling your own customized parameters. To cater to these needs, we have prepared the following guidances help you utilizing the conventional steps with our new preprocessor class. This way, you can still take advantage of the benefits of the preprocessor while also incorporating your own specific requirements.
pp = Preprocessor()
adata = dyn.sample_data.zebrafish()
pp.standardize_adata(adata, 'time', None)
|-----> Downloading data to ./data/zebrafish.h5ad
adata
AnnData object with n_obs × n_vars = 4181 × 16940
obs: 'split_id', 'sample', 'Size_Factor', 'condition', 'Cluster', 'Cell_type', 'umap_1', 'umap_2', 'batch', 'nGenes', 'nCounts', 'pMito'
var: 'nCells', 'nCounts'
uns: 'pp'
layers: 'spliced', 'unspliced'
pp.filter_cells_by_outliers(adata, max_expr_genes_s=2000)
|-----------> filtered out 244 outlier cells
AnnData object with n_obs × n_vars = 3937 × 16940
obs: 'split_id', 'sample', 'Size_Factor', 'condition', 'Cluster', 'Cell_type', 'umap_1', 'umap_2', 'batch', 'nGenes', 'nCounts', 'pMito', 'pass_basic_filter'
var: 'nCells', 'nCounts'
uns: 'pp'
layers: 'spliced', 'unspliced'
pp.filter_genes_by_outliers(adata, max_avg_exp=2000, shared_count=40)
|-----------> filtered out 13890 outlier genes
tmsb4x True
rpl8 True
ppiaa True
rpl10a True
rps4x True
...
cdc42ep1a False
camk1da False
zdhhc22 False
zgc:153681 False
mmp16b False
Name: pass_basic_filter, Length: 16940, dtype: bool
adata.var['pass_basic_filter'].sum()
3050
pp.normalize_by_cells(adata)
AnnData object with n_obs × n_vars = 3937 × 16940
obs: 'split_id', 'sample', 'Size_Factor', 'condition', 'Cluster', 'Cell_type', 'umap_1', 'umap_2', 'batch', 'nGenes', 'nCounts', 'pMito', 'pass_basic_filter', 'spliced_Size_Factor', 'initial_spliced_cell_size', 'unspliced_Size_Factor', 'initial_unspliced_cell_size', 'initial_cell_size'
var: 'nCells', 'nCounts', 'pass_basic_filter'
uns: 'pp'
layers: 'spliced', 'unspliced', 'X_spliced', 'X_unspliced'
pp.select_genes(adata, sort_by="fano_dispersion") # "cv_dispersion" or "gini"
pp.norm_method(adata) # log1p
AnnData object with n_obs × n_vars = 3937 × 16940
obs: 'split_id', 'sample', 'Size_Factor', 'condition', 'Cluster', 'Cell_type', 'umap_1', 'umap_2', 'batch', 'nGenes', 'nCounts', 'pMito', 'pass_basic_filter', 'spliced_Size_Factor', 'initial_spliced_cell_size', 'unspliced_Size_Factor', 'initial_unspliced_cell_size', 'initial_cell_size'
var: 'nCells', 'nCounts', 'pass_basic_filter', 'log_cv', 'score', 'log_m', 'frac', 'use_for_pca'
uns: 'pp', 'velocyto_SVR', 'feature_selection'
layers: 'spliced', 'unspliced', 'X_spliced', 'X_unspliced'
pp.regress_out(adata, obs_keys=['nCounts', 'pMito'])
|-----> [regress out] completed [28.5796s]
pp.pca(adata)
|-----> <insert> X_pca to obsm in AnnData Object.
AnnData object with n_obs × n_vars = 3937 × 16940
obs: 'split_id', 'sample', 'Size_Factor', 'condition', 'Cluster', 'Cell_type', 'umap_1', 'umap_2', 'batch', 'nGenes', 'nCounts', 'pMito', 'pass_basic_filter', 'spliced_Size_Factor', 'initial_spliced_cell_size', 'unspliced_Size_Factor', 'initial_unspliced_cell_size', 'initial_cell_size'
var: 'nCells', 'nCounts', 'pass_basic_filter', 'log_cv', 'score', 'log_m', 'frac', 'use_for_pca'
uns: 'pp', 'velocyto_SVR', 'feature_selection', 'PCs', 'explained_variance_ratio_', 'pca_mean'
obsm: 'X_pca'
layers: 'spliced', 'unspliced', 'X_spliced', 'X_unspliced'
dyn.tl.reduceDimension(adata, basis="pca")
dyn.pl.umap(adata, color="Cell_type", pointsize=0.2, alpha=0.4)
|-----> retrieve data for non-linear dimension reduction...
|-----> [UMAP] using X_pca with n_pca_components = 30
|-----> <insert> X_umap to obsm in AnnData Object.
|-----> [UMAP] completed [21.4056s]
|-----------> plotting with basis key=X_umap
|-----------> skip filtering Cell_type by stack threshold when stacking color because it is not a numeric type